D-セリン代謝異常と筋萎縮性側索硬化症

笹部 潤平
哺乳類の中枢神経系(特に前脳領域)には、L-セリンの1/3量程度のD-セリンが存在する。D-セリンは、グルタミン酸のイオンチャネル型受容体の一つであるN-メチル-D-アスパラギン酸(NMDA)受容体のコアゴニストとして、グルタミン酸による神経伝達に重要な働きをしていることが明らかになってきた(1)。NMDA受容体へのコアゴニストの結合は、NMDA受容体の活性に不可欠であるのみならず、グルタミン酸とNMDA受容体の親和性の増加(2)、脱感作の減少(3)、受容体の代謝促進(4)にも関与し、NMDA受容体の調節に必須の役割を担っている。このため、D-セリンは生理的に情動・認知機能・運動記憶に関与するのみならず、NMDA受容体の活動異常やグルタミン酸興奮毒性が関わるとされている精神疾患や神経疾患においても重要であると考えられてきている。
近年、運動神経疾患の一つである筋萎縮性側索硬化症(amyotrophic lateral sclerosis; ALS)において、D-アミノ酸代謝異常との関連を示唆する知見が集まってきている。ALSでは、以前よりグルタミン酸による神経興奮毒性が運動神経変性の原因の一つと考えられてきたことから、D-セリン代謝異常はNMDA受容体を介して病態へ関与するのではないかと考えられる。本稿では最近の知見を含めてALS病態生理におけるD-セリン代謝異常について概説する。
2.1.ALSとは
ALSは、最も頻度の高い成人発症の運動神経疾患である。10万人に数人の罹患率で、生涯リスクは約500人に1人である。脊髄、脳幹、大脳皮質における下位および上位運動神経細胞の選択的な変性および脱落に伴い、急速に筋萎縮および筋力低下が進行し、発症後平均3から5年という短い期間で呼吸筋麻痺にまで至ってしまう。特徴的な病理所見としては、運動神経の変性・脱落、細胞内凝集体の形成、変性した運動神経周囲のグリア細胞の活性化である。現在、唯一認可となっている治療薬は、グルタミン酸放出阻害薬(riluzole)であるが、臨床的に数ヶ月延命作用を認めるのみで治療効果に乏しい。このため、現状は主に支持療法に頼らざるを得ない状況であり、病状の進行を食い止める根治的治療法の開発が待たれている。
これまでに多くの病態仮説に基づいて治療薬シードが開発されてきたものの、臨床治験の芳しい結果は得られてこなかった。最近では、riluzoleと構造が類似するdexpramipexolが第III相試験で良好な結果を示しており期待されている(5)。
2.2.ALSの病態生理
ALSは孤発性が95%であり、5%は家族性に発症することが知られている。孤発例は因果関係が明らかな原因が同定されていないため、家族性ALSを中心にこれまで研究が進められている。家族性ALS全体の約20%を占めるCu/Zn-superoxide dismutase 1 (SOD1)の変異が最初に発見され、SOD1変異体は孤発性ALSに酷似した臨床症状をもたらす(6)。その臨床症状の類似性と変異SOD1動物モデルが典型的なALS様の表現型を有することから(7)、変異SOD1による選択的運動神経細胞死機構から孤発性ALSの病態理解が試みられてきた。酸化ストレス、ミトコンドリア機能不全、活性化グリア細胞による毒性、グルタミン酸神経興奮毒性などが運動神経変性の病態仮説として知られている(6)。現在の唯一の認可薬(riluzole)はグルタミン酸神経興奮毒性を軽減することを目的とする。
現在までに少なくとも15の家族性ALS原因遺伝子が同定されている。新たに発見されたいくつかの原因遺伝子(TAR DNA binding protein (TARDBP)、fused in sarcoma (FUS)、C9ORF72)による家族性ALSにおいて、孤発性ALSとの共通点が見い出され、TDP-43を含む細胞内凝集体が近年注目を浴びている(8,9)が、この凝集体による運動神経変性メカニズムの詳細は明らかになっていない。また、変異SOD1による家族性ALSの運動神経細胞にはTDP-43陽性の凝集体は認められず、ALS発症メカニズムの多因子性が伺える。
2.3.ALSにおける運動神経興奮毒性
ALSでは、グリア細胞におけるグルタミン酸トランスポーターの発現が脊髄前角で減少することが知られている(6)。シナプス内のグルタミン酸濃度調節は、グルタミン酸トランスポーターによって主に行われているため、ALSではシナプス内グルタミン酸濃度が上昇して神経興奮毒性を引き起こすと考えられている。神経興奮毒性はイオンチャネル型グルタミン酸受容体を介して、細胞内への過剰なCa2+流入によって起こる。イオンチャネル型グルタミン酸受容体(NMDA受容体、α-amino-3-hydroxy-5-methyL-4-isoxazolepropionic acid (AMPA)受容体、カイニン酸受容体)の中でも、Ca2+流入を阻害するGluR2サブユニットを有さないAMPA受容体が運動神経に多いことから、ALSではAMPA受容体が注目を浴びてきた(10)。一方で、NMDA受容体はCa2+透過性は高いものの脊髄には比較的発現が低いため、ほとんど注目されてこなかったが、NMDA受容体拮抗薬であるメマンチンが変異SOD1動物モデルに対して症状の進行抑制効果があることが報告され(11)、NMDA受容体と病態との関連が示唆されてきている。我々は、D-セリンが孤発性ALS患者および変異SOD1動物モデルの脊髄前角で蓄積することを発見し、ALSでの運動神経変性にNMDA受容体の活性化を介した興奮毒性も病態生理的に重要であることを示した(12)。
2.4.脊髄D-セリン調節
D-セリンは齧歯類では前脳に主に存在し、小脳以下の脳幹や脊髄では相対的に低い濃度でしか存在しない。これは合成酵素のセリンラセマーゼ(SR)が前脳に多く発現すること、分解酵素のD-アミノ酸酸化酵素(DAO)が後脳・脊髄を中心に発現することに起因する。脊髄では、生理的にはSRは脊髄後角に存在する介在神経に比較的多く発現するものの前角の神経細胞では発現に乏しい(未発表データ)。一方で、脊髄中のDAOは前索(腹側白質)および前角の腹内側のアストログリアで最も活性が高く、運動神経細胞には活性を認めない(13)。このような分布から、生理的条件下の脊髄ではD-セリンは腹側には少なく、背側脊髄に相対的に多く存在すると考えられる。尚、ヒト脊髄におけるD-セリンの分布については明らかになっていない。
生理的にはD-セリンの乏しい脊髄運動神経細胞にも、NMDA受容体の発現は認められる(14)。脊髄神経細胞のNMDA受容体のコアゴニスト部には脊髄に豊富なグリシンが結合して制御していると考えられてきたが、脊髄内でのD-セリン量が増加するG181R変異DAOマウスでは、脊髄背側神経細胞でNMDA受容体を介したシナプス伝達が増強する(15)。このことは、D-セリンはNMDA受容体への親和性がグリシンよりも高いことを示しており、圧倒的に豊富なグリシン存在下でもD-セリン量の増加はNMDA受容体を活性化へ導くと考えられる。
2.5.ALSにおけるD-セリン代謝異常
孤発性ALS患者および変異SOD1によるALS動物モデルの脊髄前角でD-セリンの蓄積を発見し、主に動物モデルを使ってその蓄積メカニズムを我々は検討してきた。変異SOD1マウスにおける脊髄D-セリン蓄積には、いくつかの要因が関与している。
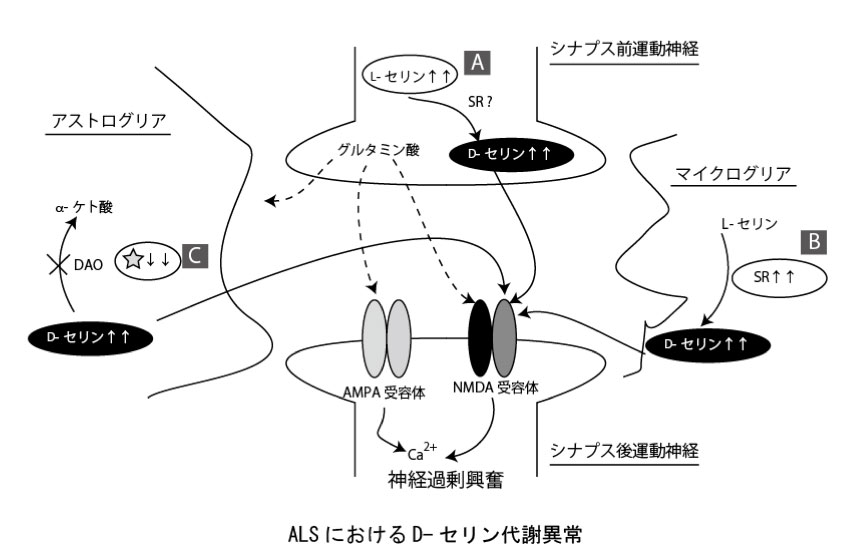
まず、第一にSRの基質となるL-セリン量の増加である。ALSモデルマウスの一つであるG37R変異SOD1マウスでは、L-セリン合成酵素である3-ホスホグリセリン酸デヒドロゲナーゼの発現が神経細胞で上昇することが知られている(16)。さらに、G93A変異SOD1マウスでは運動神経変性の発症後に脊髄中にL-セリンが増加することを我々も確認している(17)(図中A)。
第二に合成酵素であるSRの発現上昇である。SRの発現は運動神経変性の進行に伴うミクログリアの活性化に由ると考えられる。ミクログリアでは、炎症に伴いJNK/AP-1の系が活性化することでSRの発現が増加することが知られており(17)、G93A変異SOD1マウスでも、変性した運動神経細胞周囲にミクログリアが活性化し、JNKのリン酸化が亢進する(12)。実際、脊髄前角のSR陽性ミクログリアの数は、G93A変異SOD1マウスおよび孤発性ヒトALS患者脊髄で顕著に増加する(12)(図中B)。
第三に分解酵素DAOの発現および活性低下がD-セリン蓄積に寄与する。G93A変異SOD1マウスにおけるDAO活性をD-プロリンを基質とした酵素組織染色法で検出すると、網様体脊髄路に相当する脳幹網様体/脊髄前索・前角において顕著に染色性が低下する(13)。ALSの病巣ではアストログリアもミクログリアと同様に活性化することが知られているが、DAO活性は活性化アストログリアでは全く認められない(13)。脊髄全体のDAO活性は野生型の1/2程度まで低下し、mRNA・タンパク量ともに有意に減少することから(13)、正常アストログリアの減少と活性化アストログリアの増加に伴ってDAO発現が失われたものと考えられる(図中C)。
組織中D-セリン量は合成と分解のバランスによって決定されるが、DAO活性を欠損させた変異SOD1マウスを用いた検討から判断すると、上記の要因のうちDAOによる分解の低下がALSでのD-セリン蓄積に最も重要であろうと思われる(13)。
2.6.DAOの遺伝子変異と家族性ALS
DAOの活性低下は、変異SOD1マウス脊髄でのD-セリン蓄積の主たる要因であると同時に、ヒトALSでも重要であることが近年明らかとなった。
3世代6名の家族性ALS患者のゲノムスクリーニングで、DAO遺伝子のR199W変異が新規の家族性ALSの原因として同定された(18)。DAO遺伝子変異による家族性ALSは、中年発症で典型的なALS症状を示す。Arg199は種保存的なアミノ酸で、DAOのFADと基質結合部位の近傍に存在し、R199W変異はDAO活性をほぼ完全に喪失させる(13), (18)。R199Wはdominant negative変異で野生型DAOの活性を阻害すると考えられており(18)、実際、変異DAOによる家族性ALSは常染色体優性遺伝形式で発症する。
変異DAO家族性ALSでは、TDP-43陽性の凝集体を認めず、D-セリンの蓄積も含めてどのようなメカニズムで運動神経変性が起こるかは今後のさらなる検討が待たれるが、D-アミノ酸代謝異常とALS発症に密接な因果関係があることが疑われる。
2.7.D-セリン蓄積とALS病勢
脊髄中のD-セリン蓄積は運動神経変性を増悪させることがin vitroおよびin vivoの研究で示唆されている。変異SOD1マウス由来の初代培養運動神経細胞は野生型運動神経細胞と比較して、NMDA刺激に対して脆弱であり、さらにD-セリン量依存的にその脆弱性が増強する(18)。D-セリンに対する脆弱性はNR1サブユニットのD-セリン結合部位に対する阻害剤で抑制されることから(12)、D-セリン蓄積はNMDA受容体の活性化を介して運動神経細胞死を増悪すると考えられる。さらに、SRRノックアウト変異SOD1マウスでは脊髄中のD-セリン増加が抑制され、病勢の改善によって寿命が有意に延長することが近年報告された(19)。しかしながら、このマウスではALSの発症が通常の変異SOD1マウスと比較して早まるとされており(19)、D-セリン蓄積は単に運動神経変性を増悪させるのみではなく、病態へ複雑に関与している可能性が考えられる。
本稿では、D-セリン代謝異常のALSへの関与について述べた。孤発性ALSは発症や進行が様々であることから多因子疾患である可能性が高く、症状や進行による詳細な分類が必要であり、分類に応じた治療標的の開発が新規治療法の確立には不可欠であると考えられる。D-セリン代謝異常がどのようなグループのALSに関与するか、今後慎重かつ詳細な検討が進められる必要があるが、D-セリンを標的とした治療が一部のALSの病勢を抑制できるのではないかと期待している。D-セリン合成阻害や取り込み促進は、前脳の神経細胞の生理機能に大きく影響を与える可能性が高いため、後脳や脊髄に多いDAOに焦点をあててD-セリン分解を亢進させるような戦略が重要であるのではないかと考えられる。
1) Wolosker H, Dumin E, Balan L, & Foltyn VN. 2008. D-amino acids in the brain: D-serine in neurotransmission and neurodegeneration. FEBS J 275:3514-3526
2) Fadda E, Danysz W, Wroblewski JT, & Costa E. 1988. Glycine and D-serine increase the affinity of N-methyl-D-aspartate sensitive glutamate binding sites in rat brain synaptic membranes. Neuropharmacology 27:1183-1185.
3) Lerma J, Zukin RS, & Bennett MV. 1990. Glycine decreases desensitization of N-methyl-D-aspartate (NMDA) receptors expressed in Xenopus oocytes and is required for NMDA responses. Proc Natl Acad Sci U S A 87:2354-2358.
4) Nong Y, et al. 2003. Glycine binding primes NMDA receptor internalization. Nature 422:302-307.
5) Cudkowicz M, et al. 2011. The effects of dexpramipexole (KNS-760704) in individuals with amyotrophic lateral sclerosis. Nat Med 17:1652-1656.
6) Bruijn LI, Miller TM, & Cleveland DW. 2004. Unraveling the mechanisms involved in motor neuron degeneration in ALS. Annu Rev Neurosci 27:723-749.
7) Gurney ME. 1994. Transgenic-mouse model of amyotrophic lateral sclerosis. N Engl J Med 331:1721-1722.
8) Ince PG, et al. 2011. Molecular pathology and genetic advances in amyotrophic lateral sclerosis: an emerging molecular pathway and the significance of glial pathology. Acta Neuropathol 122:657-671.
9) Ludolph AC, Brettschneider J, & Weishaupt JH. 2012. Amyotrophic lateral sclerosis. Curr Opin Neurol 25:530-535.
10) Van Damme P, Dewil M, Robberecht W, & Van Den Bosch L. 2005. Excitotoxicity and amyotrophic lateral sclerosis. Neurodegener Dis 2:147-159.
11) Wang R & Zhang D. 2005. Memantine prolongs survival in an amyotrophic lateral sclerosis mouse model. Eur J Neurosci 22:2376-2380.
12) Sasabe J, et al. 2007. D-serine is a key determinant of glutamate toxicity in amyotrophic lateral sclerosis. EMBO J 26:4149-4159.
13) Sasabe J, et al. 2012. D-amino acid oxidase controls motoneuron degeneration through D-serine. Proc Natl Acad Sci U S A 109:627-632.
14) Rekling JC, Funk GD, Bayliss DA, Dong XW, & Feldman JL. 2000. Synaptic control of motoneuronal excitability. Physiol Rev 80:767-852.
15) Wake K, et al. 2001. Exaggerated responses to chronic nociceptive stimuli and enhancement of N-methyl-D-aspartate receptor-mediated synaptic transmission in mutant mice lacking D-amino-acid oxidase. Neurosci Lett 297:25-28.
16) Lobsiger CS, Boillee S, & Cleveland DW. 2007. Toxicity from different SOD1 mutants dysregulates the complement system and the neuronal regenerative response in ALS motor neurons. Proc Natl Acad Sci U S A 104:7319-7326.
17) Wu S & Barger SW. 2004. Induction of serine racemase by inflammatory stimuli is dependent on AP-1. Ann N Y Acad Sci 1035:133-146.
18) Mitchell J, et al. 2010. Familial amyotrophic lateral sclerosis is associated with a mutation in D-amino acid oxidase. Proc Natl Acad Sci U S A 107:7556-7561.
19) Thompson M, et al. 2012. Paradoxical roles of serine racemase and D-serine in the G93A mSOD1 mouse model of amyotrophic lateral sclerosis. J Neurochem 120:598-610.

笹部 潤平(ささべ じゅんぺい)氏
略 歴
1996-2002 慶應義塾大学医学部
2002-2004 慶應義塾大学病院内科学研修
2004-2008 慶應義塾大学大学院医学研究科博士課程
2008-2010 慶應義塾大学医学部解剖学特別研究助教
2010-現在 慶應義塾大学医学部解剖学助教